Tutorial: Efficiently running Migrate on clusters and other multicore machines [draft Sep 19 2012]
For an overview what MIGRATE-N does look at the MIGRATE frontpage.
MIGRATE-N is written in such a way that it is easy portable to different hardware, and that it can use many of the particular hardware speedup. Although, the current version does not use fine grained SSE2 instructions and the graphical processing unit. It allows to use multiple nodes of a computer cluster or multiple cores of a multicore CPU. The different hardware optimizations accommodate different types of option sets and data.
Typically, we distinguish between data-parallel and share-memory approaches. Data-parallel is most commonly implemented using MPI (Message Passing Interface, an open-source implementation is openmpi), MIGRATE can use this scheme to run in multiple loci and replicates at the same time in parallel (see below).
Shared-memory parallel approaches are ‘threaded’ and handle the same address space as a single-thread program (OPENMP, pthreads and GrandCentral Dispatch [on macs] are example of this). MIGRATE uses a threaded approach for heated chains and can handle them in parallel to calculate Bayes factors using thermodynamic integration or to improve mixing of the chain.
Independence of loci
Two years ago, a typical dataset for MIGRATE contained about 10 microsatellite loci, or 1 locus for DNA data. This is changing rapidly and I have seen now datasets with >10 DNA loci, and also large msat loci dataset (see an example run on 377 loci). If we assume that the loci collected are unlinked then we imply that each locus has a different coalescent history, therefore we can run them independently from each other. If the loci are a few 10,000 bases apart then because of recombination the independence assumptions works quite well. The only problem with these independent runs are how to combine them, in a maximum likelihood context this is simple, independent events are combined by multiplying the likelihood curves. In a Bayesian context this is more tricky because multiplying the n posterior distributions is incorrect because we overuse the prior n-1 times. This can be easily fixed, but without recording the prior during the run you will not be able to combine the individual loci results after the run by hand -- MIGRATE is doing this automatically. I do not know in detail how other programs are doing this exactly, but to my knowledge no other Bayesian inference in population genetics can be run in parallel (except for BEAST that can parallelize the likelihood calculation of a genealogy using the GPU, but this is rather fine-grained parallelism that is great for complex mutation models [ proteins]).
What do I mean with replicates
Replicates are common in science and commonly we would think of replicated runs of the same task or same model.
For example, assume we want to estimate parameters of a migration model then each independent locus is a replicate of an evolutionary experiment. In MIGRATE an option “replication” allows to replicate each locus. Bayesian inference will run the analysis n times and combine the collected MCMC samples (you should use this); maximum likelihood (ML)analysis allows combination of independent replicates (if you insist on ML use this; there is a second option that allows to combine over last chains).
Organization of a parallel MIGRATE run
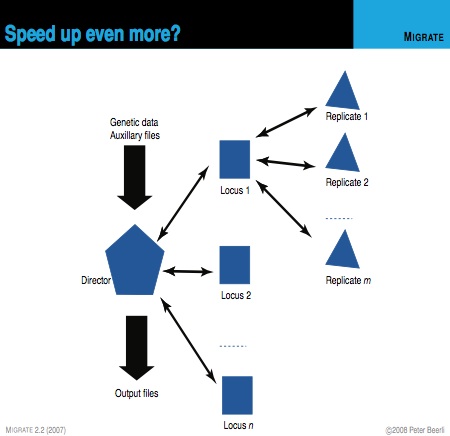
All data files (genetic data, geographic distance matrix, sample dates etc) are read by the director node. The director node schedules the worker nodes with the independent loci. Each worker communicates with the master once the locus is finished and may receive more work from the master. When replication is turned on then each locus-worker requests additional workers for replications of the current locus. The master maintains a stack of workers waiting for work and a stack for workers working as either locus-worker or replicator. The locus- worker requests a replicator and if one is available the locus-worker will communicate directly with the replicator. When the replicator has finished, it will report results to the locus - worker and also report to the master for more work. For complex problems with many loci it may help to have 10 or more replications because each MCMC chain will explore slightly different solution spaces. In 2006 I gave an Evolution meeting talk on this replication scheme. For example, if you have a dataset with 10 loci, 4 populations (16 parameters) , you may want to run 10 replicates if you have easy access to 101 computer nodes. The only problem with this approach is that you may not make your system administrator too happy because the runtime for a particular locus depends on the number of variable sites in the locus.
The combination of the replicates will not improve over a really long run, but may allow to run more samples in a restrictive environment. The backfill queue on our high-performance computing facility is such an environment. It allows a maximal job runtime of 4 hours, but we can request up to 400 computer cores. If you have a single locus mtDNA dataset and you want to sample efficiently for say a 4 population problem then you surely should sample many million steps which will result in a runtime on a single processor that may be days. The same data can be run in 100 replicates in 1/100 of the time distributed over 100 nodes, perhaps better distributed over 30 nodes. The only problems with replicates is that we need to make sure that the MCMC chain has converged in the burn-in, this will need large burn-in in because the
only few samples are taken for each replicate. Since we do not  know how long a particular run goes, I often experiment by changing the default parameters for trials run. For example instead of using the increment between collected samples (long-inc) of a 100 (default), I set it to 1, then run it over the cluster record the time (printed out as start and end in the beginning of the outfile), once I found a good combination of nodes and replicates (this means that on my backfill queue I have a total of 240 minutes before the system kills my job), therefore my test run should be done within 2.4 minutes (it may take a little longer than that because of data reading and writing). After some testing and assessment of the posterior probability histograms of the full run (you want definitely use an increment larger than 10), you will be able to run the production run. If I find that my exploration deliver unstable posteriors I will increase the number of replicates and nodes.
know how long a particular run goes, I often experiment by changing the default parameters for trials run. For example instead of using the increment between collected samples (long-inc) of a 100 (default), I set it to 1, then run it over the cluster record the time (printed out as start and end in the beginning of the outfile), once I found a good combination of nodes and replicates (this means that on my backfill queue I have a total of 240 minutes before the system kills my job), therefore my test run should be done within 2.4 minutes (it may take a little longer than that because of data reading and writing). After some testing and assessment of the posterior probability histograms of the full run (you want definitely use an increment larger than 10), you will be able to run the production run. If I find that my exploration deliver unstable posteriors I will increase the number of replicates and nodes.
Heated chains
In many situations just running replicates will be fine. But, if you want to compare models or having difficulties to reach convergence, you will need to use heating (MCMCMC -- Metropolis-coupled Markov chain Monte Carlo). MIGRATE allows to run 4 to many chains (1 cold + 4 to n heated ones). The MIGRATE source can be compiled to take into account that a CPU may have more than 1 core. On macs it uses a system called Grandcentral Dispatch and on other systems it can use pthreads. Both systems allow to run the heated chain in parallel on a single cpu/machine. When compiled for MPI on UNIX I have turned of the threading because threading will make it difficult for the cluster maintenance because the number of nodes specified for the MPI environment will not match because threading will grab available cores, thus it may impact the performance of other jobs running on the same machine. On macs there is no penalty to use parallel heated chains because the kernel is in charge of allowing the program to use all or subsets of threads or not at all. You may wonder why I did not MPI-parallelized the heated chains.
Heated chains exchange frequently information among each other, using MPIthis frequent communication among the chains that will slow down the program.
Example of effects of replicates on a single locus analysis: Running 4 populations (310 individuals) with a model of two parameters, a mutation-scaled population size and a mutation-scaled migration rate.
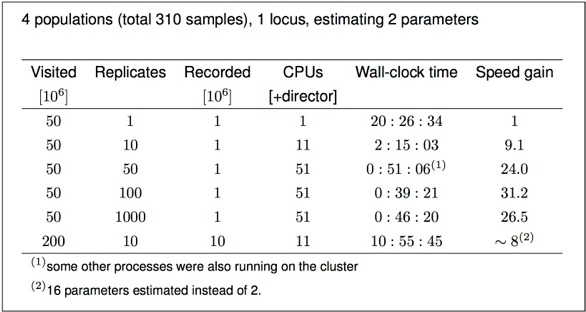
I have no “100-replicate-burn-in” plot but it will look a little worse than the “10-replicate-burn-in”.

